Protein Hydration and Interactions



The last two decades have seen an incredible revolution in structural biology: Over 90,000 protein structures have now been solved at atomic resolution. Translating this wealth of static structural information into a molecular-level understanding of their biological functions - and their impact on dynamic intracellular processes - represents a grand challenge in molecular biology, with direct implications on our understanding of human health and disease. Progress in this area hinges on our ability to understand, predict, and manipulate the interactions of various proteins, given their precise structure: Out of the menagerie of molecules that a protein encounters in the cellular milieu -- ligands, peptide fragments, nucleic acids, membranes, and other proteins -- which will it interact favorably with, how strong will the interaction be, and what are the strategies for modulating the interaction strength?
Based on recent work in our group and those of others, we hypothesize that one of the biggest challenges in accurately predicting protein interactions is precisely accounting for the role of water. Water is a key player in every biochemical interaction: Every biomolecular binding process requires replacing water-surface interactions with direct protein-protein interactions. While the direct interactions are straightforward to estimate, the protein-water interactions are difficult to quantify because proteins have incredibly complex surfaces that disrupt the inherent structure of water (strong hydrogen bonds) in countless different ways. Any hope to accurately account for the strength of water-protein interactions would require considering not only on the chemistry of the underlying protein surface, but also the precise topography.
Using principles of liquid state theory in conjunction with novel simulation techniques in explicit solvent, we are developing a computational and theoretical framework that rigorously quantifies protein-water interactions. We are applying insights from these calculations to predict the protein intermolecular interactions.
- Relevant Publications
- NB Rego, E Xi, AJ Patel. Identifying Hydrophobic Protein Patches to Inform Protein Interaction Interfaces, Proceedings of the National Academy of Sciences, 118 (6) e2018234118 (2021).
- N Rego, E Xi, AJ Patel. Protein Hydration Waters are Susceptible to Unfavorable Perturbations, Journal of the American Chemical Society, 141, 2080-2086 (2019). arXiv
- E Xi, V Venkateshwaran, L Li, N Rego, AJ Patel, and S Garde, Hydrophobicity of proteins and nanostructured solutes is governed by topographical and chemical context, Proceedings of the National Academy of Sciences, 114, 13345-13350 (2017).
- Current People
- Nick Rego
- Akash Pallath
- Lilia Escobedo
- Lizhu Zhang
Polymer Solvation and Phase Behavior
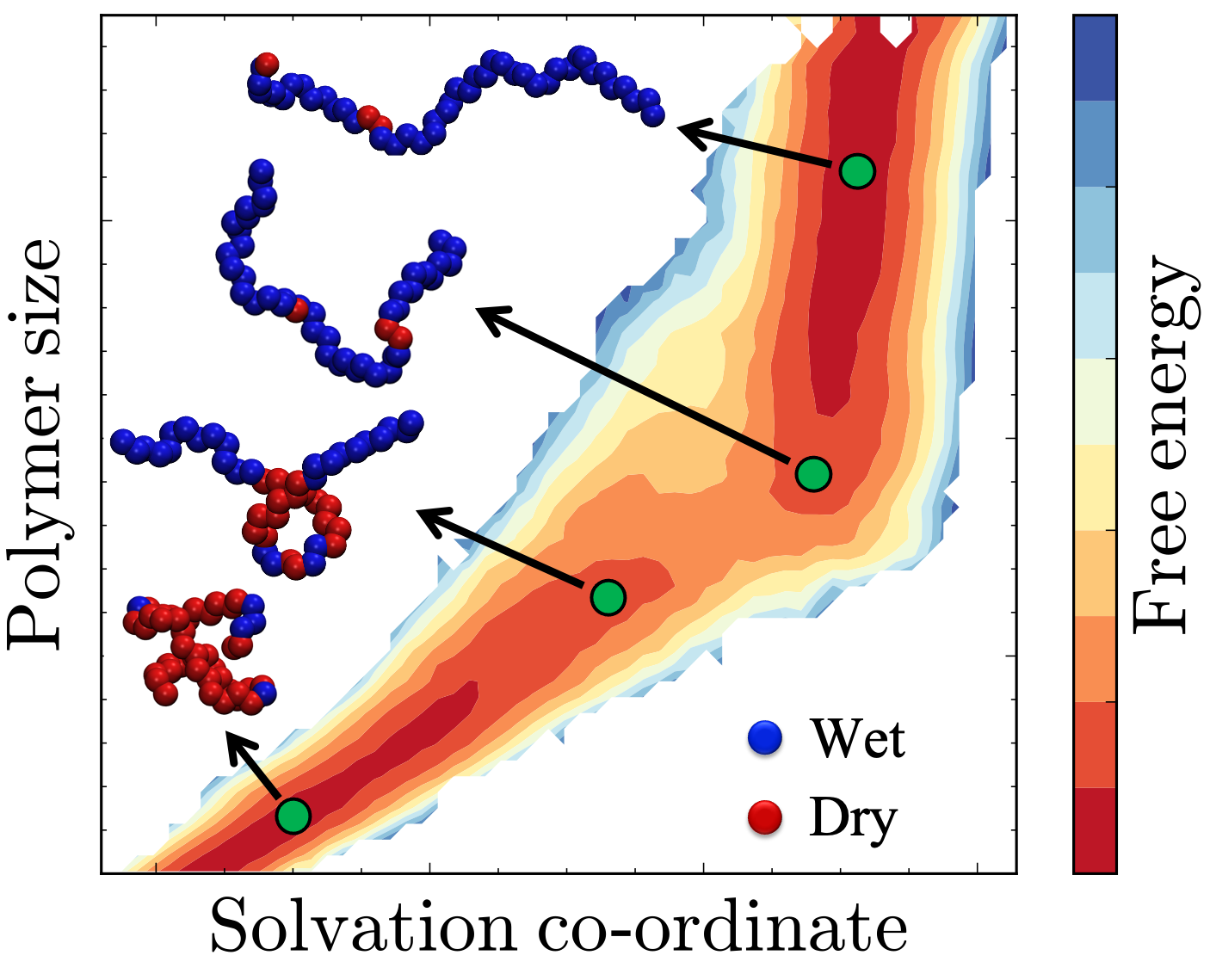
Conformationally flexible molecules, such as polymers and peptides, play an important role in a diverse soft materials and biomolecular contexts. Although flexible molecules can exist in a multitude of conformations, when dissolved in water (or other such solvents), they often display two (or more) stable basins, which are separated by free energy barriers (e.g., extended/collapsed polymers, folded/unfolded/misfolded proteins); these basins as well as transitions between them are determined, not only by the intra-molecular interactions of the solute, but also by extent to which the solute perturbs the solvent molecules in its vicinity (e.g., disruption of H-bond network in water).
By developing enhanced sampling methods for characterizing the solute conformational free energy landscape, with a strong focus on the role of solvent in mediating conformational transitions, we seek to uncover the molecular underpinnings of diverse phenomena, ranging from co-nonsolvency and stimuli-responsive polymers, to liquid-liquid phase separation in protein droplets.
- Relevant Publications
- D Dhabal, Z Jiang, A Pallath and AJ Patel. Characterizing the Interplay between Polymer Solvation and Conformation, Journal of Physical Chemistry B (2021). arXiv preprint
- Current People
- Akash Pallath
- Alex Johnson